

Because every online diet camp purports that their pet diet cures every disease known to mankind, naturally each one will give you some fantastical mechanistic story about how said diet cures type 2 diabetes mellitus (T2DM). In the paleo/keto/carnivore camp, people will often claim that T2DM is caused by chronic exposure to refined sugar, with some even going so far as to claim that it is caused by exposure to carbohydrates (CHO) more broadly (including foods like apples and even carrots). However, these days it is more trendy in that group to blame T2DM on vegetable oils, but I have previously debunked this on my blog. Over in the "whole food plant-based" (WFPB) cult, it is routinely remarked that T2DM is caused by intramyocellular lipid (IMCL), and that chronic exposure to the saturated fat (SFA) in animal products is responsible for the condition.
In this article, I'll go over the mechanistic reasoning and experiment and/or observational support for these hypotheses, as well as provide an accounting of the leading hypothesis and its supporting evidence. Let's start off by tackling the fantastical notions dreamed up by our vegan friends, and address the IMCL hypothesis.
The Intramyocellular Lipid Hypothesis
The core idea with this hypothesis is that IMCL, primarily in the form of triglycerides (TG) stored within lipid droplets in muscle cells, act through various pathways to disrupt insulin signaling. The IMCL hypothesis has drawn attention to the role of diet in modulating IMCL levels. High intakes of certain types of fats, particularly SFAs found abundantly in animal products, have been implicated by the proponents of this hypothesis in elevating IMCL levels and contributing to insulin resistance.
This association is often highlighted in discussions on the benefits of WFPB diets for reducing the risk of T2DM and improving insulin sensitivity. Advocates of WFPB diets suggest that such a diet, which is typically lower in SFA and higher in unsaturated fats like monounsaturated fats (MUFA) and polyunsaturated fats (PUFA), which can purportedly help reduce IMCL accumulation and make traction against insulin resistance.
The dietary focus is on whole grains, legumes, fruits, vegetables, nuts, and seeds, with minimal or no intake of animal products. The concern with animal products stems from their content of SFAs, dietary cholesterol, and perhaps even certain supposed inflammatory mediators (possibly Neu5Gc), which may contribute to increased IMCL levels and insulin resistance. WFPB proponents argue that replacing animal products with plant-based sources of protein and fat can mitigate these risks. But is it true?
The IMCL hypothesis attempts to highlight the potential impact of IMCL accumulation on insulin resistance and physiological markers of T2DM. While WFPB are promoted as strategies to reduce IMCL and improve metabolic health, it's important to consider the broader literature and where this hypothesis lands with respect to epistemic virtues over other, possibly more prevailing hypotheses for which there is more evidence.
The mechanistic case for the IMCL hypothesis was first outlined in the late 1990s with two small studies by Jacob, et al. and Perseghin, et al., which discovered relationships between IMCL and insulin resistance in T2DM-free subjects who were born of parents with T2DM [1][2]. In these experiments, the offspring of those with T2DM were subjected to the gold-standard measure of whole-body insulin sensitivity, the hyperinsulinemic-euglycemic clamp test (HEC). It was discovered that there was an inverse association between IMCL and insulin sensitivity, which led researchers to suspect that perhaps this biomarker was relevant to the pathophysiology of T2DM.

Fast forward to 2004, and another small study from van Loon, et al. would introduce the hypothesis' first paradox [3]. In this experiment, muscle biopsies and less precise (but still adequate) measures of insulin sensitivity were deployed in an investigation of subjects who were either sedentary, afflicted with T2DM, or who were trained athletes. The study found that the IMCL concentrations of IMCL were actually statistically significantly higher in athletic subjects compared to either sedentary or T2DM-afflicted subjects. Yet, the athletic subjects did not suffer from impaired insulin sensitivity or the hyperglycemia that is characteristic of T2DM.

Researchers Coen and Goodpaster attempted to reconcile the findings in 2012 [4]. They hypothesized that IMCL behaves differently in the context of T2DM, and that while IMCL serves as a fuel source in athletic subjects, in sedentary subjects with T2DM the IMCL seems to produce more disruptive mediators like ceramides and diacylglycerols. However, the authors tend to play fast and loose with their causal inferences, often citing animal research to buttress clear implications about what holds true in human beings. In fact, the majority of their supporting evidence is derived from mice, despite mice generally being poor facsimiles for human subjects [5].
Some of the only human research they can cite are studies wherein there was an observed association between intramyocellular ceramides (IMCC) and insulin resistance. However, there are many biomarkers that serve as proxies for insulin resistance, and there did not seem to be a clear reason proposed by the authors to favour IMCC as causal or mediating. In fact, they cite research that offered conflicting evidence with a broader sample of human subjects, showing that IMCC has no clear association with insulin sensitivity [6].
Additionally, Itani, et al (2002) discovered that the concentrations of IMCC do not differ substantially between subjects with varying degrees of insulin sensitivity [7]. In fact, these researchers challenged subjects with lipids during a HEC to try to alter lipid states in muscle tissue, and ceramides did not change. However, a legitimate criticism of this study is that the lipid challenge done using Liposyn II, which is an intravenous lipid product consisting of 50% soybean oil and 50% safflower oil, making it over half PUFA in its composition. Coen and Goodpaster also cited research on diacylglycerols, but they themselves admit that the human data isn't particularly conclusive on the matter.
To my knowledge there are no recent landmark human experiments that persuasively show that T2DM pathology can be modulated up or down with varying animal products, or even SFA, in the diet. Altogether, the hypothesis has a lot of failed predictions and phenomena to reconcile before it can be taken seriously and can even begin to be seen as epistemically virtuous compared to some other, more scientifically grounded hypotheses. What we need to see is a study that shows that removing SFA and/or animal products from the diets of those with T2DM actually makes traction against pathophysiological markers of T2DM. Ideally this would be done while also controlling for potential confounders or mediators that other competing hypotheses predict would make an impact. The aim is to demonstrate independent effects, and so far no research on the IMCL hypothesis persuasively does this.
The Sugar Hypothesis
Even though I have touched on this hypothesis five years ago [8], it bears repeating, with updated epistemic and philosophical rigour. For a recap, my original article challenges perceptions about T2DM that are common in the low carb (LC) diet sphere. I argue against the notions that sugar causes T2DM and that ketosis can somehow reverse it. But we're going to go a little deeper today. So, what is the hypothesis and how does it work? Basically, the hypothesis supposes that chronic exposure to refined sugar or insulin-stimulating CHO leads to T2DM through prolonged over-stimulation of the pancreas. This hypothesis involves several key mechanisms.
Firstly, regular intake of high-sugar or high-glycemic CHOs prompts frequent blood insulin excursions by via the pancreas. To be clear, insulin is the hormone responsible for signaling cells to absorb glucose from the bloodstream for energy or storage. The idea is that over time, constant demand for more and more insulin can lead to insulin resistance via negative feedback. This is when cells become less responsive to insulin's signals because insulin levels are too high. This requires the pancreas to produce even more insulin to achieve the same effect, placing stress on the pancreatic beta cells (which are responsible for insulin secretion).
Secondly, chronic over-stimulation of the pancreas due to persistent high sugar/CHO intake and supposed, resultant insulin resistance can lead to beta-cell dysfunction (which is characteristic of advanced T2DM). Over time, the beta cells' capacity to produce insulin can diminish due to the increased demand, oxidative stress, and subsequent glucotoxicity (toxicity due to hyperglycemia). This dysfunction contributes to the progression of T2DM, where insulin production eventually becomes insufficient to control blood sugar levels effectively.
Lastly, high levels of circulating glucose (glucotoxicity) and fatty acids (lipotoxicity) are actually detrimental to pancreatic beta cells. High glucose levels can lead to the formation of reactive oxygen species (ROS), causing an inflammatory cascade effect, damaging beta cells and impairing insulin secretion. Similarly, elevated free fatty acids (FFAs) can accumulate in non-adipose tissues, including the pancreas, causing cellular dysfunction.
The mechanisms, epidemiological, and experimental evidence for this hypothesis were most succinctly summarized by Stanhope in 2016 [9]. This review discusses the evidence and controversies surrounding the impact of sugar consumption on T2DM, including mechanisms by which excess sugar intake may promote the development of T2DM directly and indirectly. It covers the direct metabolic pathways through which fructose, a component of table sugar, sucrose, can lead to intrahepatic lipid (IHL) accumulation and decreased insulin sensitivity, contributing to the pathophysiology of T2DM independent of total caloric intake.
The author first refers to literature they themselves conducted, including three primary human trials demonstrating disturbances to energy expenditure, markers of liver function, as well as measures of IHL accumulation [10][11][12]. The first trial discovered that isocaloric feeding of fructose compared to glucose over a ten-week period significantly increased hepatic fat.

However, it's unclear what the clinical significance of this finding is, considering that the fructose-based intervention resulted in significant overfeeding and only increased fasting glucose by 5% compared to the glucose-based control. Seems like the authors tried pretty damn hard to induce a T2DM phenotype feeding fructose to subjects, and were ultimately unable to achieve that. In fact, they weren't really able to get close. Even the oral glucose tolerance test results, though far from ideal, showed an altogether normal blood glucose curve for the fructose group.

The second trial was unremarkable, and suggested that fructose consumption may decrease energy expenditure over time when isocalorically compared to glucose. Again, it's interesting, but it's far from demonstrating a cause and effect relationship between T2DM and sugar. The third trial is a bit more complicated to unpack. Basically, subjects were split into two groups, 25% of calories as either fructose or glucose as parts of eucaloric diets over 10 weeks, with the primary endpoints beings measures of liver function. Overall, the results of the trial suggest a significantly detrimental effect of fructose compared to glucose on a number of markers related to liver function. However, it's slightly more complicated than that.
While it's true that the treatment effect showed a benefit of glucose over fructose, it's also true that the marker of liver function that was negatively perturbed was gamma-glutamyl transferase (GTT). The change in GGT was also barely statistically significant compared to baseline, while markers such as aspartate aminotransferase (AST) and alanine aminotransferase (ALT) showed a non-significant decrease. Glucose showed greater reductions that were barely statistically significantly different from that of fructose.

Other than that, fructose did seem to increase uric acid levels to a statistically significant degree, making fructose a potential aggravating factor for conditions such as gout. But again, this is a far cry from causally linking fructose consumption to the development of T2DM, especially since fructose seemed to do nothing particularly interesting to markers of liver function. It is true that sugar intake is severely negatively associated with health outcomes [13]. However, while this is a consistent finding, clinical trials on sugar consistently fail to demonstrate a connection between sugar intake and the development of T2DM.
We can indirectly test the sugar hypothesis by looking at long-term data in those following ketogenic diets for T2DM remission. If subjects cut nearly all carbohydrates out of their diets, presumably this subsumes sugar, and could present a valid test of the hypothesis. One such study might be the Virta Health trial [14]. In this trial, a non-randomized, self-selected sample of eager subjects elected to participate in a cutting-edge, individualized dietary intervention program, with the aim of nutritional ketosis, called the Continuous Care Intervention (CCI), utilizing a web-app, ketone-verified adherence monitoring, and constant patient support.
Other aspects of the diet were individually prescribed to ensure safety, effectiveness, and satisfaction, including consumption of 3–5 servings of non-starchy vegetables and adequate mineral and fluid intake for the ketogenic state. At onset of dietary changes, participants were advised to consume a multivitamin, 1000–2000 IU vitamin D3, and up to 1000 mg omega-3 daily. If participants exhibited signs of magnesium depletion (e.g., muscle twitches or cramps), daily supplementation (500 mg magnesium oxide or 200 mg magnesium chloride) was suggested. If participants exhibited headaches, constipation, or lightheadedness, adequate sodium and fluid intake was recommended.
Suffice it to say, there would be little, if any, room for sugar while on such a diet. These subjects were followed up for one year, and, as expected (given the high potential for bias with such a study design), results were impressive. By the end of the first year, the control group was floundering and the CCI group managed to achieve a massive 1.3% reduction in HbA1c, which effectively pushed the once diabetic group into the non-diabetic range on average. However, this change was commensurate with a 14.3% drop in body weight, which we'll revisit in a later section of this article.
Despite these improvements, recidivism continued to climb. As of two years, the CCI group had crept back into the diabetic range on average [15]. By, 3.5 years, the CCI group was once again firmly within the diabetic range on average, having regained a significant amount of weight [16][17].

To be clear, there was a clear distinction between remission and reversal, as defined by the authors. Remission was a much more robust measure of traction against T2DM than reversal, even though it sounds like the latter is stronger than the former. Remission was defined in two parts; partial remission and complete remission. Partial remission was defined as "sub-diabetic hyperglycemia of at least 1 year duration, HbA1c level between 5.7-6.5%, without any medications (two HbA1c measurements)" and complete remission was defined as "Normoglycemia of at least 1 year duration, HbA1c below 5.7%, without any medications (two HbA1c measurements)". Reversal was defined more loosely, as "sub-diabetic hyperglycemia and normoglycemia (HbA1c below 6.5%), without medications except metformin" (Athinarayanan, et al., (2019), Supplement Table 2).
Unfortunately, all but the second year remission data is either aggregated or ambiguous, so it's difficult to make distinctions between partial and complete remission for years one and 3.5. However, at year two only 6.7% of the cohort had achieved complete remission. It's not clear what percentage of the cohort achieved complete remission by year 3.5. Furthermore, reversal rates, as they're defined, are probably just reflecting attrition rates (which were 26%), as the criteria for reversal is having achieved a sub-diabetic blood glucose at least once throughout the trial. It's actually not clear how meaningful that data even is on that definition.
While a 9% weight reduction over 3.5 years is impressive, I'll reiterate the limitations— the subjects were self-selected, and highly motivated to participate in the CCI. In fact, the patients were paying customers of Virta Health's cutting-edge primary care service. This significantly challenges the external validity of the findings, as impressive as they are, to the general population. What we're seeing are results in the context of what is likely to be extraordinary ambition to succeed, and should be interpreted with caution.
As for how these results relate to the sugar hypothesis, it's difficult to tell, had the subjects managed to control their weight. However, adherence rates to the diet dropped significantly by year two, with nutritional ketosis being achieved in 96% of CCI subjects in year one to only 14.1% by the end of year two. This non-adherence makes it difficult to infer the degree of sugar avoidance actually observed by the cohort on average, and makes cause and effect conclusions difficult to draw. There was no 3.5 year data on ketosis rates, and one can only speculate as to why. But given the poor adherence at two years, it's probably not unreasonable to assume it's because the numbers didn't look very good.
The Oxidation Hypothesis
The underlying premise is that PUFAs, particularly omega-6 fatty acids found in seed oils, are susceptible to oxidation. When oxidized, these fatty acids can form reactive compounds such as malondialdehyde (MDA) and other harmful products, leading to cellular damage and oxidative stress. In the context of the pancreas and liver, organs that, as previously mentioned, are crucial for glucose metabolism and insulin regulation, such oxidative stress could impair their function, contributing to insulin resistance and β-cell dysfunction— key features of T2DM. A tidy little bundle of sophistry indeed.
Since I've already covered how the human outcome data flies in the face of this hypothesis in a previous article [18], I won't rehash all of it here. But, essentially there is no human outcome data supporting this effect across multiple cohort studies and multiple RCTs. In the epidemiology, tight markers of seed oil consumption show a consistent inverse association with T2DM [19].
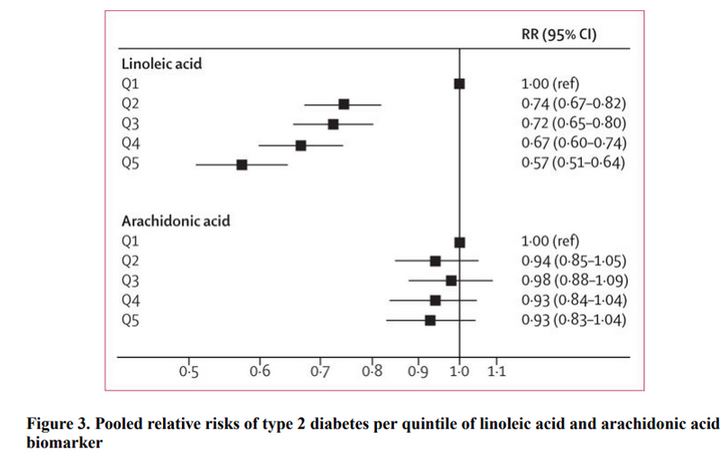
Additionally, there have been several interventions feeding large amounts of PUFA to subjects, to the exclusion of SFA, and measuring long term effects on insulin sensitivity [20][21]. In both Houtsmuller, et al. (1979) and Watts, et al. (1992), long term feeding of PUFA was associated with improvements in insulin sensitivity. In the case of the former trial, the insulin sensitivity measure, an oral glucose tolerance test (OGTT), showed improvement to glucose tolerance among diabetics over time. These results were aggregated by Hooper, et al. back in 2020 in an exploratory analysis [22].

Additionally, we can also see that Ley, et al. (2004) showed an improvement in OGTT results, but with the substitution of CHO for SFA, rather than PUFA for SFA (which is kinda funny) [23].
The Twin Cycles Hypothesis
The prevailing hypothesis, or the hypothesis that has been given the most credence in this domain, is the twin cycles hypothesis (TCH), which was spearheaded by Dr. Roy Taylor. It is closely interconnected with another concept from Dr. Taylor's work, known as the personal fat threshold (PFT). So what are these things? Let's start with the TCH. The TCH postulates that there are two stages, or "cycles", to T2DM development. The TCH was first discussed in a 2011 paper by Lim, et al. (coauthored by Dr. Taylor) detailing the development of the hypothesis from previous clinical trials performed on patients with T2DM [24]. This trial would come to be known as the Counteracting Pancreatic Inhibition by Triglyceride (CounterPoint) study, and would be reviewed in greater detail two years later by the same authors [25].
The first cycle originates in the liver, where chronic, excess calorie intake leads to ectopic fat accumulation, causing hepatic insulin resistance. The reason for this is because tissues that are energy replete will resist the action of insulin, with is a signalling hormone responsible for facilitating energy deposition in tissues. The second cycle concerns the pancreas, where the continued excess fat spills over to the pancreas (primarily via nonesterified fatty acids (NEFA), leading to lipotoxicity and subsequently impairing β-cell function. Ultimately, this contributes to β-cell dysfunction, rather than outright β-cell death, as the pivotal event in diabetes progression.
In fact, the TCH originally came about by appreciating that the assumptions typically relied upon when inferring β-cell death in T2DM patients may be faulty. Usually, we infer to β-cell death by staining pancreatic tissue for insulin; no insulin, no β-cells. However, what if the β-cells aren't really dead? What if the diabetic pancreas is just so dysfunctional that the β-cells aren't producing as much insulin? Those assumptions would have us believing that the β-cells are dead when in fact they are merely dormant due to lipotoxicity— waiting to be brought back to life upon fat mass loss. Which brings us to the next piece of the puzzle.
Now let's discuss the PFT, and how it ties into the overall picture. Essentially, the PFT posits that hepatic and pancreatic fat deposition are facilitated by accumulating more subcutaneous body fat (SBF) than one's body can tolerate, regardless of whether their body mass index (BMI) categorizes them as overweight or obese [26]. In basic terms, after non-hepatic, non-pancreatic tissues get too fat, there is a spillover of fat (precipitated by chronic calorie excess) onto the liver and the pancreas, which initiates the T2DM phenotype.
Think about it. There's just nowhere left for the energy substrates to go, be it glucose, triglycerides (TG), or even amino acids. They're all increased with T2DM, because all the tissues are energy replete. The liver and the peripheral tissues essentially play a never-ending game of ping pong with the energy substrates because no tissues are able to take on the extra calories. This hypothesis was tested with vindicating results in a recent trial by Taylor, et al. in 2024 [27]. Essentially, it seems that T2DM development and remission have very little, if anything, to do with BMI, and BMI may be a very poor risk factor for T2DM, due to the individual variation in the PFT from person to person.
Now that we have a clear understanding of the model, let's discuss the overwhelming evidence in favour of it. The clinical trials that first gave rise to the TCH were human experiments that involved both pharmaceutical and dietary means of reducing hepatic fat [28][29].
For the first study by Ravikumar, et al., a single-arm trial of 10 subjects, the drug pioglitazone was investigated for its effects on markers of T2DM, particularly postprandial endogenous glucose production (EGP) using isotope labeled glucose, as well as IHL. After 16 weeks of treatment, the pioglitazone group experienced an increase in total body weight equal to +6.2kg on average. Yet, the pioglitazone group also experienced a decrease in IHL.

However, to be fair, the pioglitazone group also experienced a significant decrease in IMCL as well, however there was no significant correlation between decreases in IMCL and EGP. There was however, a direct association between IHL and EGP. Although this trial was not controlled, it certainly doesn't produce results expected on the IMCL hypothesis, and it lends further credence to the TCH. Additionally, many markers of T2DM subsequently improved, such as plasma glucose, hemoglobin A1c (HbA1c), and even TGs, with the changes being -2.3mmol/L, -1.9%, and 0.4mmol/L, respectively.

It can also be inferred that there was a meaningful increase in insulin sensitivity, given the fact that plasma glucose dropped despite the same amount of insulin being produced by subject. Essentially, glucose disposal per unit insulin went up, implying that insulin signalling had improved.

In the second study by Petersen, et al., eight subjects with T2DM were compared to 10 healthy volunteers in a non-randomized weight loss trial over an average of seven weeks. Essentially, subjects were followed up until normoglycemia was achieved, so not every subject was subjected to the same amount of weight loss for the same time period. In this time, body weight dropped by an average of 8kg in the T2DM group. This was marked with commensurate drops in plasma glucose, plasma insulin, and TG, at -2.4mmol/L, -108pmol/L, and -0.3mmol/L, respectively.

The most interesting and surprising finding was that there was no significant change in IMCL despite weight loss. But, subjects did end up achieving normoglycemia and near normal insulin sensitivity as determined by a HEC. However, much like the previous study, insulin sensitivity was directly associated with IHL reduction. Once again, this flies in the face of the IMCL hypothesis, and offers further support for the TCH as the T2DM approached nearly normal levels of IHL. Although neither of these papers directly test for evidence of the sugar or oxidation hypotheses, it should be noted that both of these trials involve the consumption of both sugar and most likely animal products.
In light of this evidence, Steven and Taylor conducted another preliminary human trial, the CounterBalance study, involving 29 subjects in 2015 to test the effects of the same intervention in those with long- versus short-term T2DM [30]. Both groups experienced similar weight loss (short-duration: 14.8%, long-duration: 14.4%), indicating that the VLCD was effective for weight reduction regardless of diabetes duration. These results were also durable throughout the six month post-intervention follow-up.
In terms of other T2DM markers, such as plasma glucose, the response to the diet was heterogeneous among participants with long-term T2DM. Some showed similar responses to those in the short-term group and some responded slowly. By the end of the study, 87% of the short-term group and 50% of the long-term group achieved non-T2DM fasting plasma glucose levels. HbA1c levels also decreased in both groups, with a more significant reduction observed in the short-term group. However, there was an unforeseen result— about half of subjects did not respond to the treatment at all, which was not expected given nearly all previous research.

Given these results and Dr. Taylor's previous characterization of the PFT concept, the most likely hypothesis seemed clear— these people just needed to lose more weight, which we'll get to later. For now, there was enough evidence of the effectiveness of weight loss for T2DM treatment that Dr. Taylor and his colleagues decided it was time to test the efficacy of the treatment in a real-world outpatient scenario. A cluster-randomized experiment was designed and undertaken, and would come to be known as the Diabetes Remission Clinical Trial (DiRECT) [31].

The DiRECT trial involved 298 subjects across 49 primary care practices (or clusters) randomized two groups, a control group receiving the standard of care for T2DM management, and the treatment group receiving a three stage program: stage one, total diet replacement; stage two, food reintroduction; phase three, weight maintenance. For the first stage, the treatment group received a liquid diet consisting of four packets of meal replacement formula, which totaled around 825-853 kcal/day for three to five months (depending on patient-specific goals). For the second stage, after subjects completed the first weight loss phase of the trial, food was gradually reintroduced over a period of two weeks to two months. During the last stage, patients were followed up over the course of around a year. The results were encouraging.
Over the course of the trial, the treatment group lost an average of 10kg, with over 86% of them achieving T2DM remission by the end of the first year. An interesting finding was that on average, at the end of the first year, the treatment group still technically qualified as obese, despite the vast majority of them achieving T2DM remission. This would come to be the first nail in the coffin with respect to the idea that T2DM was somehow coupled to BMI. Additionally, patients experienced significant improvements to quality of life without serious side effects or complications. Altogether the treatment was successful, well-tolerated, and produced impressive rates of T2DM remission that was durable long-term.
However, Dr. Taylor's group published two follow-up, long term durability studies on the DiRECT trial [32][33]. The results of these follow-up studies was bitter-sweet. At the two-year follow-up, only 41% of the treatment group remained in remission compared to the previous year, and only 10% at five years of follow-up. These results were not promising for the treatment over time once people were reintroduced to their previous diets and left without practitioner support. Even after the two-year follow-up, 94 or the remaining 101 subjects in the treatment group were given access to extended care, which only resulted in 13% remission rates within the extended care group at five years.
It sounds bleak, but let's think about it. Dr. Taylor's research was essentially testing two things at the end of the day— the conceptual model of T2DM, encompassing the TCH and the PFT, as well as the efficacy of radical weight loss in an outpatient setting. With respect to the former, Taylor's work has been a resounding success, and it buttresses the strongest model of T2DM development and progression that we currently have. In regards to the latter, radical supervised weight loss with the practitioner support did not yield terribly promising results beyond two years. All isn't lost, though.
The important thing is that we now have a firm grasp about what causes T2DM. It isn't sugar, seed oils, animal fat, or any other harebrained dietary boogeyman. It's just energy poisoning, with a simple, easy-to-understand etiology; if you gain more fat than your body can tolerate, you develop the phenotype of T2DM. If at that point you do indeed lose enough body fat to fall back below the maximum your body can withstand, T2DM remission follows. The last piece of the puzzle is figuring out what factors cause over-consumption, and how to durably address excessive body fat.
Discussion
In conclusion, while a myriad of hypotheses continue to circulate within nutritional and diabetic research spheres regarding the genesis and treatment of T2DM, it becomes increasingly clear that simplicity often guides the best practice. The TCH, underscored by the PFT, offers a cogent framework suggesting that T2DM is not merely a product of specific dietary components like sugars, SFA, or PUFA, but rather a complex interplay of caloric excess leading to dysfunctional energy storage and insulin response. As emerging studies, such as those by Dr. Taylor and his colleagues, indicate, interventions aiming at substantial and sustained weight loss may hold the key to reversing T2DM, provided these interventions are maintained with consistent medical oversight and support.
While the exploration of dietary influence on T2DM remains important, we also have to acknowledge the apparently lack of elasticity of our food culture and the stark tendency toward recidivism with dietary intervenions. Emerging pharmacological interventions, particularly GLP-1 receptor agonists, are presenting promising frontiers in the management and potential reversal of this condition. As we advance, the integration of such pharmaceutical approaches alongside dietary management could revolutionize the treatment landscape of T2DM. Emphasizing the synergy between medication and lifestyle changes will likely be pivotal in crafting effective, personalized treatment plans that address both the biochemical and lifestyle facets of diabetes care.
Thanks for reading! If you enjoy my writing and want more content like this, consider pledging to my Patreon!
References:
[1] Jacob, S., et al. “Association of Increased Intramyocellular Lipid Content with Insulin Resistance in Lean Nondiabetic Offspring of Type 2 Diabetic Subjects.” Diabetes, vol. 48, no. 5, May 1999, pp. 1113–19. PubMed, https://doi.org/10.2337/diabetes.48.5.1113.
[2] Perseghin, G., et al. “Intramyocellular Triglyceride Content Is a Determinant of in Vivo Insulin Resistance in Humans: A 1H-13C Nuclear Magnetic Resonance Spectroscopy Assessment in Offspring of Type 2 Diabetic Parents.” Diabetes, vol. 48, no. 8, Aug. 1999, pp. 1600–06. PubMed, https://doi.org/10.2337/diabetes.48.8.1600.
[3] van Loon, Luc J. C., et al. “Intramyocellular Lipid Content in Type 2 Diabetes Patients Compared with Overweight Sedentary Men and Highly Trained Endurance Athletes.” American Journal of Physiology. Endocrinology and Metabolism, vol. 287, no. 3, Sept. 2004, pp. E558-565. PubMed, https://doi.org/10.1152/ajpendo.00464.2003.
[4] Coen, Paul M., and Bret H. Goodpaster. “Role of Intramyocelluar Lipids in Human Health.” Trends in Endocrinology and Metabolism: TEM, vol. 23, no. 8, Aug. 2012, pp. 391–98. PubMed, https://doi.org/10.1016/j.tem.2012.05.009.
[5] Leenaars, Cathalijn H. C., et al. “Animal to Human Translation: A Systematic Scoping Review of Reported Concordance Rates.” Journal of Translational Medicine, vol. 17, no. 1, July 2019, p. 223. PubMed, https://doi.org/10.1186/s12967-019-1976-2.
[6] Skovbro, M., et al. “Human Skeletal Muscle Ceramide Content Is Not a Major Factor in Muscle Insulin Sensitivity.” Diabetologia, vol. 51, no. 7, July 2008, pp. 1253–60. PubMed, https://doi.org/10.1007/s00125-008-1014-z.
[7] Itani, Samar I., et al. “Lipid-Induced Insulin Resistance in Human Muscle Is Associated with Changes in Diacylglycerol, Protein Kinase C, and IkappaB-Alpha.” Diabetes, vol. 51, no. 7, July 2002, pp. 2005–11. PubMed, https://doi.org/10.2337/diabetes.51.7.2005.
[8] “The Nutrivore: Sugar Doesn’t Cause Diabetes, and Ketosis Doesn’t Reverse It.” The Nutrivore, 16 Oct. 2019, https://thenutrivore.blogspot.com/2019/10/sugar-doesnt-cause-diabetes-and-ketosis.html.
[9] Stanhope, Kimber L. “Sugar Consumption, Metabolic Disease and Obesity: The State of the Controversy.” Critical Reviews in Clinical Laboratory Sciences, vol. 53, no. 1, 2016, pp. 52–67. PubMed, https://doi.org/10.3109/10408363.2015.1084990.
[10] Stanhope, Kimber L., et al. “Consuming Fructose-Sweetened, Not Glucose-Sweetened, Beverages Increases Visceral Adiposity and Lipids and Decreases Insulin Sensitivity in Overweight/Obese Humans.” The Journal of Clinical Investigation, vol. 119, no. 5, May 2009, pp. 1322–34. PubMed, https://doi.org/10.1172/JCI37385.
[11] Cox, C. L., et al. “Consumption of Fructose-Sweetened Beverages for 10 Weeks Reduces Net Fat Oxidation and Energy Expenditure in Overweight/Obese Men and Women.” European Journal of Clinical Nutrition, vol. 66, no. 2, Feb. 2012, pp. 201–08. PubMed, https://doi.org/10.1038/ejcn.2011.159.
[12] Cox, Chad L., et al. “Consumption of Fructose- but Not Glucose-Sweetened Beverages for 10 Weeks Increases Circulating Concentrations of Uric Acid, Retinol Binding Protein-4, and Gamma-Glutamyl Transferase Activity in Overweight/Obese Humans.” Nutrition & Metabolism, vol. 9, no. 1, July 2012, p. 68. PubMed, https://doi.org/10.1186/1743-7075-9-68.
[13] Meng, Yantong, et al. “Sugar- and Artificially Sweetened Beverages Consumption Linked to Type 2 Diabetes, Cardiovascular Diseases, and All-Cause Mortality: A Systematic Review and Dose-Response Meta-Analysis of Prospective Cohort Studies.” Nutrients, vol. 13, no. 8, July 2021, p. 2636. PubMed, https://doi.org/10.3390/nu13082636.
[14] Hallberg, Sarah J., et al. “Effectiveness and Safety of a Novel Care Model for the Management of Type 2 Diabetes at 1 Year: An Open-Label, Non-Randomized, Controlled Study.” Diabetes Therapy: Research, Treatment and Education of Diabetes and Related Disorders, vol. 9, no. 2, Apr. 2018, pp. 583–612. PubMed, https://doi.org/10.1007/s13300-018-0373-9.
[15] Athinarayanan, Shaminie J., et al. “Long-Term Effects of a Novel Continuous Remote Care Intervention Including Nutritional Ketosis for the Management of Type 2 Diabetes: A 2-Year Non-Randomized Clinical Trial.” Frontiers in Endocrinology, vol. 10, 2019, p. 348. PubMed, https://doi.org/10.3389/fendo.2019.00348.
[16] Veazie, S., Vela, K., & Helfand, M. (2020). Evidence Brief: Virtual Diet Programs for Diabetes. Evidence Synthesis Program (ESP), Portland VA Health Care System, VA Health Services Research & Development Service, Washington, DC. Available at: http://www.hsrd.research.va.gov/publications/esp/
[17] McKenzie, Amy, et al. “SUN-LB113 A Continuous Remote Care Intervention Utilizing Carbohydrate Restriction Including Nutritional Ketosis Improves Markers of Metabolic Risk and Reduces Diabetes Medication Use in Patients With Type 2 Diabetes Over 3.5 Years.” Journal of the Endocrine Society, vol. 4, no. Suppl 1, May 2020, p. SUN-LB113. PubMed Central, https://doi.org/10.1210/jendso/bvaa046.2302.
[18] Hiebert, Nick. “A Comprehensive Rebuttal to Seed Oil Sophistry.” The Nutrivore, 1 Nov. 2021, https://www.the-nutrivore.com/post/a-comprehensive-rebuttal-to-seed-oil-sophistry.
[19] Wu, Jason H. Y., et al. “Omega-6 Fatty Acid Biomarkers and Incident Type 2 Diabetes: Pooled Analysis of Individual-Level Data for 39 740 Adults from 20 Prospective Cohort Studies.” The Lancet. Diabetes & Endocrinology, vol. 5, no. 12, Dec. 2017, pp. 965–74. PubMed, https://doi.org/10.1016/S2213-8587(17)30307-8.
[20] Houtsmuller, A. J., et al. “Favorable Influences of Linoleic Acid on the Progression of Diabetic Micro- and Macroangiopathy in Adult Onset Diabetes Mellitus.” Progress in Lipid Research, vol. 20, Jan. 1981, pp. 377–86. ScienceDirect, https://doi.org/10.1016/0163-7827(81)90070-9.
[21] Watts, G. F., et al. “Effects on Coronary Artery Disease of Lipid-Lowering Diet, or Diet plus Cholestyramine, in the St Thomas’ Atherosclerosis Regression Study (STARS).” Lancet (London, England), vol. 339, no. 8793, Mar. 1992, pp. 563–69. PubMed, https://doi.org/10.1016/0140-6736(92)90863-x.
[22] Hooper, Lee, et al. “Reduction in Saturated Fat Intake for Cardiovascular Disease.” The Cochrane Database of Systematic Reviews, vol. 5, no. 5, May 2020, p. CD011737. PubMed, https://doi.org/10.1002/14651858.CD011737.pub2.
[23] Ley, Sarah J., et al. “Long-Term Effects of a Reduced Fat Diet Intervention on Cardiovascular Disease Risk Factors in Individuals with Glucose Intolerance.” Diabetes Research and Clinical Practice, vol. 63, no. 2, Feb. 2004, pp. 103–12. PubMed, https://doi.org/10.1016/j.diabres.2003.09.001.
[24] Lim, E. L., et al. “Reversal of Type 2 Diabetes: Normalisation of Beta Cell Function in Association with Decreased Pancreas and Liver Triacylglycerol.” Diabetologia, vol. 54, no. 10, Oct. 2011, pp. 2506–14. PubMed, https://doi.org/10.1007/s00125-011-2204-7.
[25] Taylor, R. “Banting Memorial Lecture 2012: Reversing the Twin Cycles of Type 2 Diabetes.” Diabetic Medicine: A Journal of the British Diabetic Association, vol. 30, no. 3, Mar. 2013, pp. 267–75. PubMed, https://doi.org/10.1111/dme.12039.
[26] Taylor, Roy, and Rury R. Holman. “Normal Weight Individuals Who Develop Type 2 Diabetes: The Personal Fat Threshold.” Clinical Science (London, England: 1979), vol. 128, no. 7, Apr. 2015, pp. 405–10. PubMed, https://doi.org/10.1042/CS20140553.
[27] Taylor, Roy, et al. “Aetiology of Type 2 Diabetes in People with a ‘normal’ Body Mass Index: Testing the Personal Fat Threshold Hypothesis.” Clinical Science (London, England: 1979), vol. 137, no. 16, Aug. 2023, pp. 1333–46. PubMed, https://doi.org/10.1042/CS20230586.
[28] Ravikumar, Balasubramanian, et al. “Pioglitazone Decreases Fasting and Postprandial Endogenous Glucose Production in Proportion to Decrease in Hepatic Triglyceride Content.” Diabetes, vol. 57, no. 9, Sept. 2008, pp. 2288–95. PubMed, https://doi.org/10.2337/db07-1828.
[29] Petersen, Kitt Falk, et al. “Reversal of Nonalcoholic Hepatic Steatosis, Hepatic Insulin Resistance, and Hyperglycemia by Moderate Weight Reduction in Patients with Type 2 Diabetes.” Diabetes, vol. 54, no. 3, Mar. 2005, pp. 603–08. PubMed, https://doi.org/10.2337/diabetes.54.3.603.
[30] Steven, S., and R. Taylor. “Restoring Normoglycaemia by Use of a Very Low Calorie Diet in Long- and Short-Duration Type 2 Diabetes.” Diabetic Medicine: A Journal of the British Diabetic Association, vol. 32, no. 9, Sept. 2015, pp. 1149–55. PubMed, https://doi.org/10.1111/dme.12722.
[31] Lean, Michael Ej, et al. “Primary Care-Led Weight Management for Remission of Type 2 Diabetes (DiRECT): An Open-Label, Cluster-Randomised Trial.” Lancet (London, England), vol. 391, no. 10120, Feb. 2018, pp. 541–51. PubMed, https://doi.org/10.1016/S0140-6736(17)33102-1.
[32] Lean, Michael E. J., et al. “Durability of a Primary Care-Led Weight-Management Intervention for Remission of Type 2 Diabetes: 2-Year Results of the DiRECT Open-Label, Cluster-Randomised Trial.” The Lancet. Diabetes & Endocrinology, vol. 7, no. 5, May 2019, pp. 344–55. PubMed, https://doi.org/10.1016/S2213-8587(19)30068-3.
[33] Lean, Michael Ej, et al. “5-Year Follow-up of the Randomised Diabetes Remission Clinical Trial (DiRECT) of Continued Support for Weight Loss Maintenance in the UK: An Extension Study.” The Lancet. Diabetes & Endocrinology, vol. 12, no. 4, Apr. 2024, pp. 233–46. PubMed, https://doi.org/10.1016/S2213-8587(23)00385-6.